[ ]:
!pip install rdkit wget
Molecular Descriptors¶

[2]:
import itertools
import numpy as np
import matplotlib.pyplot as plt
import rdkit.Chem as Chem
from rdkit.Chem import Descriptors
from rdkit.Chem.rdmolops import GetAdjacencyMatrix
from IPython.display import SVG
Load From SMILES¶
[3]:
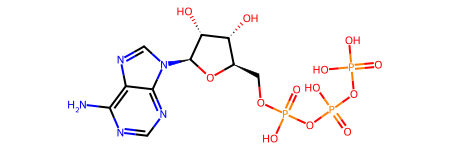
smi = "C1=NC(=C2C(=N1)N(C=N2)[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(O)OP(=O)(O)OP(=O)(O)O)O)O)N"
mol = Chem.MolFromSmiles(smi)
mol
[3]:
Molecular Properties¶
RDKit provides interfaces for many advanced molecular properties, e.g. Molecular Weight, Hydrogen bonding donors, Hydrogen bonding acceptors and number of rings. rdkit provides an interface called Descriptors.CalcMolDescriptors to calculate all molecular descriptors available in the package.
[4]:
# quantitative estimation of drug-likeness (QED)
q = Descriptors.qed(mol)
print("QED:", q)
QED: 0.1974220337838805
[ ]:
## Download source code to compute synthetic availability score from rdkit
!python -m wget https://raw.githubusercontent.com/rdkit/rdkit/master/Contrib/SA_Score/sascorer.py
!python -m wget https://raw.githubusercontent.com/rdkit/rdkit/master/Contrib/SA_Score/fpscores.pkl.gz
[6]:
import os, sys
from rdkit.Chem import RDConfig
# using literature contributors
# from https://github.com/rdkit/rdkit/tree/master/Contrib
sys.path.append(os.path.join(RDConfig.RDContribDir, 'SA_Score'))
import sascorer
s = sascorer.calculateScore(mol)
print("Synthetic Availability:", s)
Synthetic Availability: 4.41361201850629
Functional Groups¶
SMARTS is used to define a molecular pattern, such as funtional groups. We use Chem.MolFromSmarts to define a pattern, and use GetSubstructMatches to return atom groups in the molecule that match the pattern. GetSubstructMatches returns a tuple of tuples.
References:
[7]:
# define a pattern
primary_amine = "[NX3;H2;!$(NC=[!#6]);!$(NC#[!#6])][#6]"
pattern = Chem.MolFromSmarts(primary_amine)
# match the patter
hits = mol.GetSubstructMatches(pattern)
print(f"Number of amine functional group in ATP: {len(hits)}")
print(f"Atom indices in each selected functional group: {hits}")
Number of amine functional group in ATP: 1
Atom indices in each selected functional group: ((30, 2),)
[8]:
atom_ids = list(itertools.chain(*hits))
print(f"Atoms matching the functional group: {atom_ids}")
atoms = [] # get all atoms
for a in mol.GetAtoms():
atoms.append(a.GetIdx())
bond_ids = []
for bond in mol.GetBonds():
aid1 = atoms[bond.GetBeginAtomIdx()]
aid2 = atoms[bond.GetEndAtomIdx()]
for hit in hits:
# make sure bonds connecting atoms in the same functional group
if (aid1 in hit) and (aid2 in hit):
bond_ids.append(mol.GetBondBetweenAtoms(aid1, aid2).GetIdx())
break
print(f"Bonds matching the functional group: {bond_ids}")
Atoms matching the functional group: [30, 2]
Bonds matching the functional group: [29]
[9]:
def draw_single_mol(mol, size=(300, 300), **highlights):
# copy the molecule to avoid modifying molecule
mol = Chem.Mol(mol)
drawer = Chem.Draw.rdMolDraw2D.MolDraw2DSVG(*size)
if highlights is not None:
Chem.Draw.rdMolDraw2D.PrepareAndDrawMolecule(drawer, mol, **highlights)
else:
drawer.DrawMolecule(mol)
drawer.FinishDrawing()
svg = drawer.GetDrawingText()
return svg.replace('svg:','')
[10]:
SVG(
draw_single_mol(mol, highlightAtoms=atom_ids, highlightBonds=bond_ids)
)
[10]:

Molecular Graph¶
[11]:
def mol_with_atom_index(mol):
mol = Chem.Mol(mol)
for atom in mol.GetAtoms():
atom.SetAtomMapNum(atom.GetIdx())
return mol
SVG(draw_single_mol(mol_with_atom_index(mol)))
[11]:

[12]:
adjacency_matrix = GetAdjacencyMatrix(mol)
print("Shape of Adjacency matrix:", adjacency_matrix.shape)
## uncomment the following command to print the adjacency matrix,
# and check a few adjacency_matrix[i][j] with the atom indices indicated above
adjacency_matrix
Shape of Adjacency matrix: (31, 31)
[12]:
array([[0, 1, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[1, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 1, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 1],
[0, 0, 1, 0, 1, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 1, 0, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[1, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 1, 0, 0, 1, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 1, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 1, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 1, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 1, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 1, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 1, 1, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 1, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 1, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 1, 1, 1, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 1, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 1,
1, 1, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0,
0, 0, 1, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 1, 0, 1, 1, 1, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 1, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 1, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 1, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0]], dtype=int32)
[13]:
allowed_atom_types = ["C", "N", "O", "P"]
# create onehot embeddings
# dim+1 to deal with unknown types
atom_emb = np.zeros((len(mol.GetAtoms()), len(allowed_atom_types)+1))
for atom in mol.GetAtoms():
try:
atom_idx = atom.GetIdx()
ele = atom.GetSymbol()
type_idx = allowed_atom_types.index(ele)
atom_emb[atom_idx, type_idx] = 1
except:
atom_emb[atom_idx, -1] = 1
[14]:
atom_emb
[14]:
array([[1., 0., 0., 0., 0.],
[0., 1., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[0., 1., 0., 0., 0.],
[0., 1., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[0., 1., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[0., 0., 1., 0., 0.],
[1., 0., 0., 0., 0.],
[0., 0., 1., 0., 0.],
[0., 0., 0., 1., 0.],
[0., 0., 1., 0., 0.],
[0., 0., 1., 0., 0.],
[0., 0., 1., 0., 0.],
[0., 0., 0., 1., 0.],
[0., 0., 1., 0., 0.],
[0., 0., 1., 0., 0.],
[0., 0., 1., 0., 0.],
[0., 0., 0., 1., 0.],
[0., 0., 1., 0., 0.],
[0., 0., 1., 0., 0.],
[0., 0., 1., 0., 0.],
[0., 0., 1., 0., 0.],
[0., 0., 1., 0., 0.],
[0., 1., 0., 0., 0.]])
[15]:
allowed_bond_types = [Chem.rdchem.BondType.SINGLE, Chem.rdchem.BondType.DOUBLE, \
Chem.rdchem.BondType.TRIPLE, Chem.rdchem.BondType.AROMATIC]
# create onehot embeddings
# dim+1 to deal with unknown types
bond_emb = np.zeros((len(mol.GetBonds()), len(allowed_bond_types)+1))
for bond in mol.GetBonds():
try:
bond_idx = bond.GetIdx()
b_type = bond.GetBondType()
type_idx = allowed_bond_types.index(b_type)
bond_emb[bond_idx, type_idx] = 1
except:
bond_emb[bond_idx, -1] = 1
[16]:
bond_emb
[16]:
array([[0., 0., 0., 1., 0.],
[0., 0., 0., 1., 0.],
[0., 0., 0., 1., 0.],
[0., 0., 0., 1., 0.],
[0., 0., 0., 1., 0.],
[0., 0., 0., 1., 0.],
[0., 0., 0., 1., 0.],
[0., 0., 0., 1., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[0., 1., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[0., 1., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[0., 1., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[1., 0., 0., 0., 0.],
[0., 0., 0., 1., 0.],
[0., 0., 0., 1., 0.],
[1., 0., 0., 0., 0.]])